目的
- atom indexを一意に振り直す方法を解説する
- CanonicalRankAtomsやRenumberAtoms関数を理解する
はじめに
さまざまなデータベースから分子構造を入手したり自分でMolオブジェクトを作成していると、同じ分子においてもatom indexの順番が異なることが多々あると思います。
SMILESからMolオブジェクトを作成するだけなら、SMILES→Molオブジェクト→SMILESの操作でカノニカルなSMILESが入手できるので困ることはありません。
しかし、3次元構造が決定しているなどの理由でMolオブジェクトのままatom indexを振り直したい場面があると思います。自分はこのような場面に遭遇したので、atom indexを一意に振り直す方法をまとめようと思いました。
コード
以下のRDKit cookbookを参考に実装しました。
前提
N-メチルアセトアミドを例として実装します。
前提として、2種類のSMILES, CC(=O)NCとCC(NC)=OからMolオブジェクトを作成するとatom indexは以下のようになります。
from rdkit import Chem
from rdkit.Chem import Draw
# 順番が異なる2つSMILESからMolオブジェクトを作成
smiles1= "CC(=O)NC"
smiles2= "CC(NC)=O"
mol1= Chem.MolFromSmiles(smiles1)
mol2= Chem.MolFromSmiles(smiles2)
# atomLabelプロパティにatom indexを設定
for mol in [mol1,mol2]:
for atom in mol.GetAtoms():
atom.SetProp("atomLabel", str(atom.GetIdx()))
# mol1(左)とmol2(右)を描写
Draw.MolsToGridImage([mol1,mol2], molsPerRow=2, subImgSize=(200,200))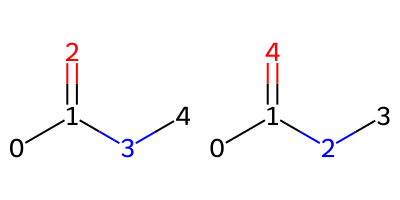
このように、同一の化合物においてもatom indexが異なってしまうことが多々あります。
以下では、この2種類のMolオブジェクト(N-メチルアセトアミド)のatom indexをカノニカルに振り直します。
Reorder Atoms[実装]
- Chem.CanonicalRankAtoms(mol)関数でカノニカルなatom indexを取得
- Chem.RenumberAtoms(mol, newOrder)関数でatom indexを振り直す
# canonicalなindexを取得
neworder1= tuple(zip(*sorted([(j, i) for i, j in enumerate(Chem.CanonicalRankAtoms(mol1))])))[1]
neworder2 = tuple(zip(*sorted([(j, i) for i, j in enumerate(Chem.CanonicalRankAtoms(mol2))])))[1]
# canonicalなatom indexを振り直す
mol1_new = Chem.RenumberAtoms(mol1, neworder1)
mol2_new = Chem.RenumberAtoms(mol2, neworder2)
# atomLabelプロパティにatom indexを設定
for mol in [mol1_new,mol2_new]:
for atom in mol.GetAtoms():
atom.SetProp("atomLabel", str(atom.GetIdx()))
# mol1_new(左)とmol2_new(右)を描写
Draw.MolsToGridImage([mol1_new,mol2_new], molsPerRow=2, subImgSize=(200,200))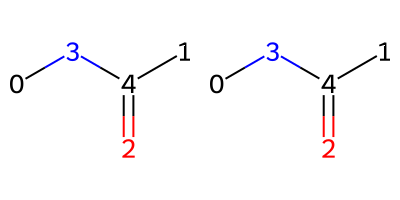
Chem.RenumberAtoms(mol, newOrder) について
Chem.RenumberAtomsの引数は、Molオブジェクトと新しい順番に整列したatom indexのリストです。
新しいatom indexのリストではないことに注意。
また、戻り値として新しいMolオブジェクトを返すので、元のMolオブジェクトは変化しません。
mol_old= Chem.MolFromSmiles("CC(=O)NC")
mol_new= Chem.RenumberAtoms(mol_old, [3,2,4,1,0])
# atomLabelプロパティにatom indexを設定
for mol in [mol_old,mol_new]:
for atom in mol.GetAtoms():
atom.SetProp("atomLabel", str(atom.GetIdx()))
# mol_old(左)とmol_new(右)を描写
Draw.MolsToGridImage([mol_old,mol_new], molsPerRow=2, subImgSize=(200,200))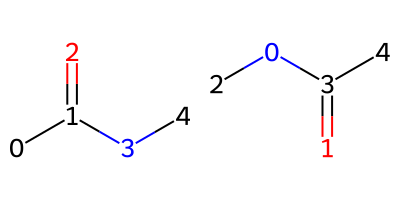
回転してわかりにくいですが、newOrder=[3,2,4,1,0]を引数とすると、元の[0,1,2,3,4]というatom indexが[4,3,1,0,2]に変化しました。つまり、newOrderの[3,2,4,1,0]は元のindex 3→0, 2→1と振り直すことを表します。そのため、上記の実装でtuple(zip(*sorted([(j, i) for i, j in enumerate(Chem.CanonicalRankAtoms(mol1))])))[1]のような複雑な処理が行われています。
終わり。